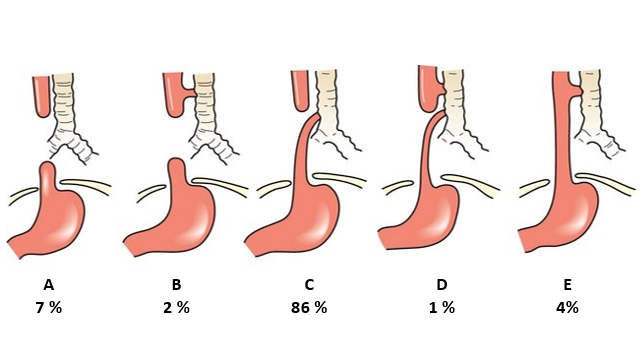
Definizione: L’atresia esofagea (AE) con o senza fistola esofago-tracheale (FET) è una malattia rara, anche se è la più comune anomalia congenita dell’esofago. Si verifica ogni 3-5000 nascite. È causata da una errata differenziazione dell’esofago e della trachea (che durante lo sviluppo dell’embrione e del feto derivano da un unico “intestino primitivo”), la cui causa non è ancora completamente nota. Ne consegue una separazione dell’esofago in due monconi, uno superiore che viene dalla bocca e uno inferiore che si collega con lo stomaco, che possono anche essere molto distanti (long-gap atresia) e creare problemi di ricostruzione chirurgica (Tipo A). A questo si associano spesso comunicazioni (fistole) tra trachea e moncone esofageo prossimale, distale o entrambi (Tipo B, C, D). A volte, infine, la canalizzazione esofagea è conservata, ed è presente solo una comunicazione tra esofago e trachea (cosiddetta “fistola ad H”, Tipo E). Nella figura sono illustrate queste varianti, secondo la classificazione in uso (Gross, 1953), con le rispettive frequenze.
ATRESIA ESOFAGEA
Diagnosi: La diagnosi viene fatta in genere alla nascita, anche se può essere sospettata durante la gravidanza con l’ecografia (assenza di aria nello stomaco e presenza di polidramnios, cioè un eccessivo accumulo di liquido amniotico nell’utero). Alla nascita, il segno principale di AE è la impossibilità di far progredire un catetere introdotto dalla bocca in esofago per più di una decina di centimetri. Altri segni clinici che si possono manifestare nelle ore successive sono una abbondante salivazione, ma soprattutto episodi di soffocamento con colorito bluastro della pelle (cianosi) durante l’allattamento. La conferma diagnostica si può ottenere con una semplice radiografia del torace utilizzando come contrasto la presenza di aria nella sacca dell’esofago prossimale, evitando così la somministrazione di mezzo di contrasto con conseguente passaggio nelle vie aeree. Una tracheo-broncospia (cioè l’osservazione diretta con l’endoscopio della trachea e dei bronchi) perfeziona la diagnosi.
Purtroppo, sino al 50% dei casi, possono essere presenti altre anomalie associate: sono soprattutto a carico del cuore (tetralogia di Fallot), dell’apparato muscolo-scheletrico, dell’intestino in genere o della sua ultima parte (ano e retto), oltre che dell’apparato urinario e genitale. L’associazione di alcune di queste anomalie costituisce vari gruppi di manifestazioni (o sindromi), come la sindrome VACTERL (anomalie Vertebrali, Anali, Cardiache, Tracheali, Esofagee, Renali e degli arti – Limb), o altre. Tali anomalie associate influiscono grandemente sul tipo di trattamento e la sua possibilità di successo.
Trattamento: L’AE è una condizione incompatibile con la vita e richiede un intervento chirurgico urgente ma non emergente, da eseguire nelle prime ore di vita del piccolo paziente: il neonato deve essere quindi trasportato al più presto presso un Centro di riferimento. Infatti, data la complessità dei piccoli pazienti e l’entità dell’intervento, che viene eseguito attraverso una apertura del torace, è opportuno che questo venga eseguito presso un Centro di riferimento con adeguata esperienza (viene considerato come adeguato un Centro che esegua almeno 5 di tali interventi all’anno).
In genere è possibile ricostruire l’esofago abboccando chirurgicamente tra loro i due monconi superiore e inferiore (anastomosi), oltre che effettuando la chiusura chirurgica della comunicazione tra esofago e trachea. Qualche volta, specialmente se i due monconi sono molto distanti (> 3 cm, long-gap atresia) e non riunificabili tra loro, è necessario ricostruire l’esofago interponendo un altro organo, come lo stomaco, o un tratto di intestino (colon).
Le complicanze dell’intervento possono essere un ritardo di guarigione della anastomosi (fistole) o un restringimento della stessa, che raramente richiedono nuovo intervento ma possono essere gestite con trattamento conservativo nel primo caso, o tramite “allargamenti” (dilatazioni) per via endoscopica nel secondo. Anche un reflusso gastro-esofageo, con episodi di vomito dopo la poppata e possibili problemi respiratori (tosse e broncopolmoniti da inalazione), oltre che un ritardo di crescita, può frequentemente manifestarsi sin dai primi giorni o settimane dall’intervento.
Fortunatamente, negli ultimi decenni si è assistito ad un progressivo miglioramento della sopravvivenza dei piccoli pazienti, che ora arriva al 95% (negli anni ‘50 e ’60 del secolo scorso era del 65%). Ciò è stato possibile certamente grazie ai progressi delle tecniche e dei materiali chirurgici, ma anche e soprattutto a quelli della terapia intensiva e della anestesia neonatale, del supporto ventilatorio e nutrizionale, della terapia antibiotica. Inoltre, è da tutti ormai accettato che l’intervento deve essere eseguito il più precocemente possibile. La mortalità è oggi in pratica limitata solo ai neonati con gravi anomalie associate, di per sé potenzialmente letali.
Follow-up a lungo termine: Sempre più bambini operati per AE alla nascita raggiungono l’adolescenza e l’età adulta. Superato l’intervento chirurgico, negli anni successimi diventano importanti gli aspetti nutrizionali e gastroenterologici, quelli legati allo sviluppo neurologico e dell’accrescimento in generale. Anche gli aspetti psicologici non vanno sottovalutati. In generale, comunque, tali pazienti mantengono una eccellente qualità di vita, simile a quella di soggetti normali. Tuttavia, la valutazione a lungo termine rivela che continua ad essere presente una serie di disturbi. La difficoltà a deglutire (disfagia) è molto comune e può arrivare sino all’85% dei pazienti operati. Tale difficoltà può essere legata ad un restringimento infiammatorio della sutura tra moncone esofageo prossimale e distale (stenosi cicatriziale), ma anche ad un alterato controllo neurologico dell’esofago malformato, già presente alla nascita, che comporta la perdita della capacità di spingere verso lo stomaco il cibo ingerito, con difficoltà di alimentazione, frequenti rigurgiti e sintomi respiratori (tosse, broncopolmoniti) che possono persistere nell’età adulta.
Sintomi e complicanze causati da un reflusso gastroesofageo (cioè dalla risalita di materiale gastrico acido verso l’esofago) sono estremamente frequenti, e possono arrivare al 60% dei pazienti, alcuni dei quali, se non trattati, possono sviluppare da adulti un esofago di Barrett. Se è presente un grave reflusso sarà necessario instaurare un trattamento con farmaci (omeprazolo e derivati) per lungo tempo, se non per tutta la vita, mentre un discreto numero può anche richiedere un intervento chirurgico antireflusso.
Nella minoranza dei pazienti in cui non è stato possibile eseguire una ricostruzione diretta dell’esofago e quindi si trovano con l’esofago sostituito da un altro organo come lo stomaco o il colon, si possono manifestare altre complicanze specifiche, come la difficoltà dello stomaco a svuotarsi nell’intestino o un allungamento e inginocchiamento del pezzo di colon usato, che assume aspetto “a calzetta”, con conseguente frequente rigurgito, difficoltà ad alimentarsi e problemi respiratori.
Tutto questo sottolinea l’importanza di seguire nel tempo questi pazienti in modo multidisciplinare e con indagini strumentali cadenzate come l’esofagogastroduodenoscopia (EGDS) ed altre. Recentemente ERNICA (Esophageal Reference Network for rare Inherited and Congenital Anomalies, https://ern-ernica.eu), che si occupa attivamente a livello Europeo di AE oltre che di altre rare malformazioni congenite gastro-intestinali) ha pubblicato delle linee guida per seguire a lungo termine questi pazienti, con un programma multidisciplinare che coinvolga chirurghi, gastroenterologi, pneumologi, nutrizionisti, psicologi e altri, sia di ambito pediatrico che dell’adulto. In particolare, le linee guida consigliano di eseguire diverse esofagogastroduodenoscopie: la prima ad un anno di età, seguita da almeno altre due sino all’età di transizione all’età adulta, per poi seguire i protocolli di sorveglianza endoscopica tipici dell’adulto (una EGDS ogni 3-5 anni).
La fase della transizione verso l’età adulta è di particolare importanza. Presuppone un cambiamento totale della gestione del malato, che deve imparare a gestire in modo attivo la sua malattia, assumendone in prima persona il controllo e la responsabilità, prima a carico dei genitori, che ora devono fare un passo indietro. Anche il ruolo del medico deve cambiare, da un approccio essenzialmente orientato alla famiglia con alto grado di coinvolgimento dei genitori, tipico del pediatra, ad un rapporto binario paziente-medico. Quest’ultimo deve conoscere bene la malattia e le sue possibili complicanze a lungo termine, per poterle individuare precocemente e, se possibile, trattarle. Non sempre tutto questo è facile, ma in questi ultimi anni c’è stata una generale presa di coscienza del problema della transizione, con conseguente creazione di ambulatori e servizi dedicati, almeno nei grossi Centri di riferimento.
Appare infine fondamentale il coinvolgimento il più precoce possibile dei pazienti e dei loro genitori nelle associazioni di sostegno di pazienti e familiari, come ad esempio questa associazione ALMA, o anche la FATE (Associazione Famiglie con ATresia Esofagea, https://www.atresiaesofagea.it), dove trovare risposte adeguate ai vari problemi che una rara malattia come l’AE può determinare nella vita dei soggetti che ne sono affetti.
 Prof Mario Costantini
Prof Mario Costantini
Dipartimento di Scienze Chirurgiche e Oncologiche
Università di Padova